")

For years, diagnosing cardiac amyloidosis has been regarded as an academic pursuit in many regions, primarily due to limited treatment options. However, following major scientific breakthroughs and the approval of effective therapies like Tafamidis and Daratumumab in multiple countries, the outlook for patients with cardiac amyloidosis has significantly improved. Patients worldwide can now benefit from timely diagnosis and treatment, shifting the focus from mere symptom management to improving long-term outcomes.
With these advancements, clinicians are increasingly considering cardiac amyloidosis in patients with unexplained or hard-to-manage heart failure. Early recognition is critical because the symptoms of cardiac amyloidosis are often subtle or easily mistaken for more common conditions, leading to delayed diagnosis. If left untreated, the disease can progress with a potentially poor prognosis. Timely intervention with the right therapies, however, has been shown to improve survival rates and quality of life, underscoring the importance of making this diagnosis sooner rather than later.
Amyloidosis – a disease of protein misfolding
In normal health, your body produces proteins that are folded precisely to carry out normal functions. Eventually, these proteins are degraded and recycled. Amyloidosis describes a group of diseases in which an abnormal protein misfolds and then aggregates to form resistant strands (or fibrils) which are difficult for the body to break down.
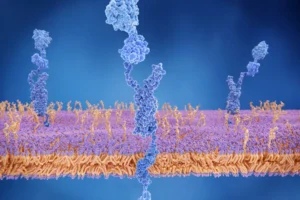
These fibrils are subsequently deposited in organs causing dysfunction, depending on where the amyloid fibrils are deposited – i.e. the heart, nerves, kidney, gastrointestinal tract – different symptoms occur. For instance, when amyloid proteins deposit in the heart, it thickens the walls of the heart, the stiffened heart cannot relax reducing the amount of blood that can be pumped, patients often feel shortness of breath, severe fatigue and experience leg swelling.
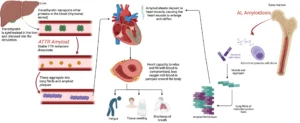
Amyloidosis is classified according to the protein which misfolds. For example, AL amyloidosis is caused by the aggregation of abnormal light-chain proteins (a component of antibodies) while ATTR amyloidosis is caused from the aggregation of transthyretin (a transport protein produced by the liver).
The most common types of amyloidosis, are ATTR (Transthyretin) and AL (Light-chain) amyloidosis. While estimates vary, between 160-300 cases of AL amyloidosis are diagnosed each year in Australia, and ATTR is estimated to occur in 6-7% of patients with heart failure with preserved ejection fraction (1, 2).
A challenging diagnosis
The diagnosis of amyloidosis is often delayed, as the symptoms produced may be vague and can mimic more commonly encountered medical conditions. Common symptoms include:
- Feeling short of breath
- Swelling in your ankles or feet
- Fainting
- Numbness or burning in your hands or feet
- Frothiness of your urine with a high protein concentration
The most common scenarios in which amyloidosis should be considered are:
- A stiff or thickened heart leading to unexplained heart failure (often seen on heart ultrasound scans)
- The loss of large amount of protein in the urine, also called nephrotic syndrome
- Numbness, tingling or pain in the hands or feet
- Difficulty regulating your blood pressure between lying and standing, leading to light-headedness and falls
Amyloidosis may be recognized on the basis of a patients presenting symptoms, the appearance of the heart on echocardiogram, or findings from blood or urine testing. After the syndrome is recognized, the diagnosis is confirmed through a combination of laboratory and radiological studies. In some cases, a biopsy (or tissue sampling) is required. A bone scan called a technetium pyrophosphate scan, can diagnose most cases of ATTR amyloidosis. A bone marrow biopsy is required for the diagnosis of AL amyloidosis.
New Treatments for Amyloidosis
Amyloidosis is a complex condition caused by the abnormal buildup of amyloid proteins in various organs, which can lead to severe organ dysfunction. The impact on a patient’s health largely depends on the specific organs affected—commonly the heart, kidneys, liver, nervous system, and gastrointestinal tract—and the extent of the damage. Early symptoms are often non-specific, such as fatigue or weight loss, making early detection challenging. For patients with advanced organ involvement, especially with cardiac amyloidosis, the condition can become life-threatening. Hence, prompt diagnosis and immediate treatment initiation are crucial to slowing disease progression and stabilizing organ function.
Goals of Amyloidosis Treatment
The primary objective of amyloidosis treatment is to prevent further amyloid protein deposits from accumulating in the organs. By halting amyloid plaque formation, treatment allows the body’s natural processes to potentially reverse some of the existing damage and improve organ function. Treatment approaches depend on the amyloid type and involve targeted therapies that either reduce the production of amyloid proteins or help clear them from the body.
Treatment Options Available in Australia
In Australia, three main therapies are approved and available for specific types of amyloidosis:
Tafamidis (for ATTR Cardiac Amyloidosis)
Tafamidis is approved for treating transthyretin amyloidosis (ATTR) that affects the heart. ATTR amyloidosis results from the misfolding of transthyretin, a protein produced in the liver, which then accumulates in the heart. Tafamidis works by stabilizing the transthyretin protein, slowing its breakdown and reducing amyloid deposition in the heart. Clinical trials have shown that Tafamidis can improve both survival and quality of life for patients with cardiac ATTR amyloidosis.
Patisiran (for Hereditary ATTR Amyloidosis)
For hereditary ATTR amyloidosis, where amyloid protein builds up in peripheral nerves and sometimes the heart, Patisiran is an RNA interference (RNAi) therapy that targets the liver to reduce transthyretin production. By lowering transthyretin levels in the blood, Patisiran helps to decrease amyloid deposition in affected tissues, thereby alleviating nerve and other organ-related symptoms.
Daratumumab (for AL Amyloidosis)
AL amyloidosis, another common type, is associated with the production of abnormal light chains by plasma cells in the bone marrow. In this case, the accumulation primarily impacts the heart and kidneys. Daratumumab, an antibody used in combination with chemotherapy, targets and depletes the abnormal plasma cells, reducing the production of the amyloidogenic light chains. This combination therapy has shown to be effective in stabilizing or even improving organ function in patients with AL amyloidosis.
Ongoing Research and Clinical Trials
Research in amyloidosis is rapidly advancing, with several clinical trials currently exploring new therapeutic options, including potential gene therapies and novel small-molecule stabilizers. For patients with amyloidosis, especially those who may not respond fully to standard treatments, participation in clinical trials offers access to innovative therapies. For information on these trials and their availability, patients and healthcare providers can refer to the Australian Amyloidosis Network (AAN) website, where a comprehensive list of clinical trials and the respective centers conducting them is maintained: Australian Amyloidosis Network – Clinical Trials.
Early intervention in amyloidosis is essential, as treatments can stabilize the disease and may improve survival and quality of life. With advancements in therapies and clinical trials, the outlook for amyloidosis patients continues to improve.
Final thoughts
The recognition of amyloidosis as a significant cause of heart failure has grown as research has illuminated the complex mechanisms and specific organ damage associated with the condition. For years, amyloidosis was frequently underdiagnosed, with its symptoms often mistaken for other forms of heart disease, such as hypertensive heart disease or restrictive cardiomyopathy. However, the advent of new targeted therapies—like Tafamidis, Patisiran, and Daratumumab—has shifted the landscape, encouraging healthcare providers to re-evaluate diagnoses in patients with unexplained heart failure or progressive organ dysfunction. Improved diagnostic imaging techniques, biomarker testing, and genetic analysis now make it possible to identify amyloidosis earlier and more accurately, which is crucial for timely treatment initiation.
Patients have a pivotal role in this evolving landscape. By actively monitoring and reporting symptoms, particularly any new or unusual signs such as rapid fatigue, unexplained weight loss, numbness in extremities, or gastrointestinal changes, patients can aid clinicians in identifying amyloidosis earlier. Awareness and self-advocacy are critical, as symptoms may develop gradually and overlap with other conditions, leading to potential delays in diagnosis.
The Australian Amyloidosis Network (AAN) offers a valuable resource for patient education and support, including information on symptoms, available treatments, and local and international clinical trials. The AAN website (AAN) serves as a central point for patients to access reliable, comprehensive information on amyloidosis and its management, empowering them to seek informed care and participate actively in their health decisions.
Conclusion
In conclusion, advancements in the understanding and treatment of amyloidosis have transformed it from a once-overlooked diagnosis to a treatable condition with promising outcomes. Both healthcare professionals and patients play a role in this transformation: clinicians by embracing new diagnostic and therapeutic approaches and patients by being vigilant and proactive in recognizing and reporting symptoms. This collaborative approach is essential in ensuring that more patients receive a timely diagnosis and access to effective treatments that can significantly improve their quality of life and health outcomes.
References:
- AbouEzzeddine OF, Davies DR, Scott CG, Fayyaz AU, Askew JW, McKie PM, et al. Prevalence of Transthyretin Amyloid Cardiomyopathy in Heart Failure With Preserved Ejection Fraction. JAMA Cardiology. 2021;6(11):1267-74.
- Wisniowski B, McLeod DSA, Adams R, Harvey Y, Brown I, McGuire L, et al. The epidemiology of amyloidosis in Queensland, Australia. Br J Haematol. 2019;186(6):829-36.